EV71 or Adenovirus? Hypothesis Arising from Two Epidemics Spanning 1997/98
by
A. Introduction
In the middle of 1997, a viral outbreak of cardiopulmonary failure (mislabeled as a Coxsackie epidemic) in Sarawak killed over 30 young children. While the first cases were reported from Sibu, it spread rapidly to Kuching. The deaths were riding on the crest of an epidemic of Hand-foot-and-mouth disease (HFMD). In the summer months of the following year, a HFMD epidemic of historic proportion ravaged Taiwan, killing at least 78 young children with very similar clinical presentations to those of the Sarawak outbreak. Among the baffling array of probable viruses that have emerged, enterovirus 71 and a new strain of adenovirus have surfaced in the spate of scientific papers generated from the two epidemics.
My own interest in the epidemics was sparked off by witnessing the futile attempts of three doctors to resuscitate a two-year old child brought into the emergency room in a state of cardiogenic shock in April 1997, no doubt one of the earlier victims of the Sarawak outbreak, even before anyone realized we had an epidemic on hand. In order to 'make sense' out of the published evidence, I have made some bold postulations, which are also attempts at mind-reading what protagonists in the field would be thinking about. The article is written by a non-virologist for the general readership of the Newsletter. I hope my elementary understanding of virology and epidemiology will allow readers to grasp the significance of the data or technique involved and to make their own conclusions. Readers who wish to pursue issues and data mentioned with greater vigour are advised to read the original publications.
B. Clinical and Epidemiological Features: Comparing the Two Epidemics
The first three cases of the Sarawak viral outbreak was described by the pediatricians S. Krishnan, Wong See Chang (1) and Tan Poh Tin (2) as being admitted in compensated cardiogenic shock after a short febrile illness with tachycardia (180 - 200/min.) and tachypnea. Fluid replacement led to findings of lung crepitations (pulmonary oedema) and echocardiogram images of "poorly contractile globular left ventricle." Two children who died later demonstrated acute flaccid paralysis (AFP) and one had convulsions.
The 34 children who died in the epidemic within the following 5 months were aged between 5 months to 7 years with a mean age of 20 months. All deteriorated rapidly in spite of intubation and other supportive measures, dying within 29 hours after admission (1).
CNS involvement was better described by the Taiwanese doctors. LY Chang and his co-authors of Tauyuan's Chang Gung Childrens' Hospital summarized the CNS pathologies as aseptic meningitis, encephalitis, poliomyelitis-like syndrome or encephalomyelitis (3). Huang and colleagues, in another Taiwanese paper, described neurological symptomatology as myoclonus, tremor, cranial nerve palsies, ataxia and acute flaccid paralysis. They classified the severity of the neurological involvements into three grades of 'rhombencephalitis', with the highest fatality rate among the Grade III. Five out of seven children classified as Grade III died of "rapid cardiorespiratory failure" (4).
The Sarawakian children affected had similar CNS manifestation, but less severe than those suffered by Taiwanese children. While only 10 of these cases have CSF available for laboratory analysis, 9 of these showed signs of aseptic meningitis (CSF showed pleocytosis with negative bacterial culture) (1).
For those who care to read the papers, the similarity in clinical presentations of the two outbreaks are quite obvious. The dissimilarity was in differing perceptions of the pathogenesis of death. While Cardosa and colleagues summarized the clinical condition as "cardiorespiratory failure", "severely depressed myocardial contractility" and "cardiogenic shock" (1), the Taiwanese used terms like "pulmonary edema or pulmonary hemorrhage and myocarditis" (3, 5). The Taiwanese doctors, to various extent, refer to the putative pathophysiological connections between the brain and the cardiopulmonary systems by invoking the term "neurogenic pulmonary edema" (3,4,5,6). Chang, Huang and Lin were excited enough about a 8-year-old girl with "fulminant neurogenic pulmonary edema with HFMD" that prompted them to sent a research letter giving full clinical details of the case to the Lancet (6).
On epidemiological grounds, the similarities of the Taiwan epidemic with the Sarawakian one were also striking. Both series of outbreak deaths rode on the crests of much larger waves of mainly benign HFMD. Soon after the outbreak announcement in Sarawak, the State Health Department held a meeting with almost all the general practitioners practising in Sibu - the epicentre of the epidemic. By an impromptu tally of numbers provided by the GPs, the cumulative number of HFMD seen in Sibu in the past month added up to 10,000. A sentinel clinic system was then set up mainly to collect clinical samples for virological investigations. In Taiwan, 850 sentinel-clinics were set up in all 22 cities and countries, comprising 8.7% of the doctor population. They reported a total of 129,106 HFMD cases in two waves between March 29 till the end of the year. Projecting this figure to what would have been seen by all the 9427 outpatient doctors practising throughout the island, the total number of patients could have been a staggering 1,483,977 (5).
The dissimilarity lies in the proportion of fatal cases. Taiwan officially reported 78 fatalities (5) while the Sarawakian deaths were close to of 34 (1). Even allowing for considerable error in official figures due to underreporting (of up to 5 times the official figure), the Taiwan deaths seems disproportionately low when measured against the gargantuan proportion of the HFMD epidemic.
C. The hunt for the killer virus
While some viruses may be fiendishly difficult to culture and identify, it is even more difficult to prove cause-and-effect for epidemic deaths (Koch's postulate being usually not fulfilled). The obvious question to ask is, "What is the primary organ failure that leads to death ?" Because viruses are known to have differing affinity for different tissues/organs (virus-tropism) the answer to the question may help to establish the culpability of the killer. In the case of EV71 vs Adenovirus, both can be neurotoxic; but adenoviruses have the edge over EV71 if deaths were related to myocardial mechanisms. Bowles, Towbin and associates, using PCR and other molecular techniques since about ten years' ago, have put forward the view that adenovirus is an important cause of myocarditis and dilated myocardiopathy (DMC) (7, 8, 9,10).
Shortly after the arrival of the young epidemiologist Jim Alexander Jr, head of a team of 3 experts from Atlanta's Center for Disease Control, the controversy started. Local doctors were told that their perception of acute heart failure and cardiogenic shock as the main cause of epidemic deaths were askew, it was probably EV71-associated brain stem damage which killed the children. The local scientist and doctors were taken aback at the brash confidence of the CDC expert. His point of view became more transparent when the local doctors came across a paper of which he was the first author (11). In the paper, which summed up the result of 14 years of CDC's study of outbreaks of EV71 in the United States, the authors concluded that EV71 had proved to be an increasing important and virulent pathogen in viral outbreaks. They were of the opinion that that it might have been underdiagnosed because not all laboratories were sufficiently equipped to culture and identify it (11).
CDC made the first identification of enterovirus 71 from 2 cultures of the 13 cultures grown from samples of Sarawakian patients. This result was first announced by Professor Lam Sai Kit of the Medical Microbiology Department in University of Malaya. The same University of Malaya team also successfully cultured the enterovirus subsequently from brain tissue from University Hospital’s epidemic patients. The virus was identified as EV71 by either immunoflourescence with monoclonal antibody or RT-PCR (reverse transcriptase- polymerase chain reaction) (12,13). The crux of the evidence was reported in a paper in Journal of Pediatrics, describing 4 young children who died of cardiopulmonary collapse with minimal neurological manifestations during the secondary outbreak in Peninsular Malaysia that followed closely on the heels of the Sarawak outbreak. Post mortem revealed extensive damage to the medulla and pons which also yielded positive culture of EV71. A postulation of neurogenic pulmonary edema (NPE) was made (12), citing Baker (1957) who had linked sudden pulmonary edema with bulbar poliomyelitis damage to the vasomotor centers in the medulla (14).
The Malaysian paper was widely cited by most of the Taiwanese authors because it anteceded their paper and espoused the same views on etiology and pathogenesis, ie. EV71 infection and damage of the hindbrain of children cause rapid death by way of neurogenic pulmonary edema and cardiac dysfunction.
There is dominance of evidence and opinion towards EV71 in all the Taiwanese publications. The main paper describing the epidemic, by Monto Ho and others, showed that 81 % of the 96 severe enteroviral infections yielded EV71 isolates. Furthermore, "enterovirus 71 was isolated from all 25 patients with encephalitis and pulmonary edema or hemorrhage". In comparison, of the 11 patients with aseptic meningitis, only 5 had EV71 while 6 had other enteroviruses. As for those who died in the outbreak, most of the death (83%) were due to pulmonary edema or hemorrhage. Thirty-two out of 33 patients who died from pulmonary edema had positive culture for EV71, only one had Coxsackie B5 (5).
Among the milder cases, other than EV71 which is the most frequent virus isolated from inpatients (61.9%) and outpatients (48.7%), Coxsackie A16 also featured prominently. This last mentioned enterovirus was isolated from 27.5% of inpatients and 37.2% of outpatients. The other enteroviruses identified among patients were untyped viruses; coxsackieviruses A24, B1, B2, B3 and B5; echoviruses 6, 7, 11, 22 and 27; as well as polio virus (5).
The alternative candidate for the culprit virus in the Malaysian epidemic, a new strain of adenovirus (subgenus B), was cultured in Professor Jane Cardosa's laboratory, UNIMAS and was named temporarily as Sibu97 (1). While there was news of such a discovery from the Sarawak epidemic in cyberspace and a press conference a full five months after the EV71 announcement, not much account was given to it by the 'Final Bulletin' of the "Coxsackie Epidemic" by the Ministry of Health. Even the main paper entitled "Isolation of subgenus B adenovirus during a fatal outbreak of enterovirus 71-associated hand, foot, and mouth disease in Sibu, Sarawak" was forwarded to Lancet as late as two years after the epidemic. Unlike its Taiwan counterparts, where many authors have collaborated on large papers published in leading journals, this is the only major peer-reviewed paper on the Sarawak epidemic.
In the Cardosa et al paper (1), the main virological evidence came from 16 of the 20 outbreak cases who died in Sibu Hospital and eight surviving cases who had AFP. The new adenovirus was identified in 10 of the dead victims (63%) and 5 of 8 surviving cases (63%). The new virus was difficult to culture (fastidious): multiple passages had to be made with the A549 cells before any cytopathic effect was seen. Conditions for annealing in PCR were also modified. In comparison, enteroviruses were found in only 3 (19%) of the dead victims and in none of the surviving cases. Two of the enteroviruses cultured were EV71 and one was a ECHO 25. Furthermore, at least five of the cases (one third of those with virus isolated) had what looked like a double infection where Sibu97 co-existed with another virus, which could be a EV71, ECHO 25, dengue, or herpes.
D. Insights Provided by Molecular Virology
Molecular techniques were employed extensively in the discovery of the subgenus B adenovirus by Jane Cardosa. Cloning and sequencing of the DNA nucleotides of its hexon gene and comparison with databases of virus sequences (GenBank, NCBI, USA) showed up the 8% of uniqueness of its nucleotide sequences from known members of the adenovirus genus, the closest member being Type 16 (1). After two years of sequencing and comparison with known genotypes, Cardosa indicated that it was most probably a new strain of Adenovirus 21 (15).
AbuBakar et al (16) subjected the 5' untranslated region (5'UTR) of 13 EV71 isolates to partial sequencing and classified their genomic relationship in two clusters. The Peninisular isolates were found in both clusters while the Sarawak isolates were only found in the II cluster. The nucleotide sequences from isolates derived from fatal cases had at least 13 nucleotide position differences with previously reported EV71 5'UTR sequences. The authors concluded that there were at least two different potentially virulent EV71 co-circulating in Malaysia during the 1997 HFMD outbreak.
The most comprehensive study of the molecular evolution of EV71 must surely come from the CDC, who have been working on the problem for nearly 30 years. The culmination of their labour was published by Betty Brown et al in a paper entitled, "Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998" (17). In this paper, Mark Pallansch and his team (plus an Australian collaborator) described the analysis of the complete 891 nucleotide sequence of VP1, one of the four proteins that have 60 copies to form a viral capsid, in all the EV71 collected in 29 years. The comparison of the nucleotide sequences allowed them to plot a phylogenetic tree of all the strains and even estimate the evolutionary rate in terms of nucleotide substitutions per year. The comparison demonstrated three distinct EV71 'genotypes', named as A, B and C. While all strains of the three genotypes were identical by at least 94% of their deduced amino acid sequence, the three genotypes differ in nucleotide sequences by 16.5% to 19.7%. The A genotype was represented by a single strain isolated in California in 1970 from an encephalitis case and designated as BrCr-CA-70. The B genotype's 'most primitive' strain (in the phylogenetic tree) was isolated from Columbia in 1994. The later strains of genotype C probably originated from the same stock as a 1985 Hubei, China strain designated as 0667-CHN-85 (18); these evolved into the recent strains found in Australia, US and the rest of the world. Strains which were close to each other genetically were further grouped into "clusters". Both genotypes B and C have two clusters each.
The EV 71 strains from the 1997 Sarawak outbreak was classified within the B genotype while the Peninsular strains had two (genotype) B and several C strains (17, 19). Furthermore, the last mentioned was at some distance, in fact a different 'cluster', from the 1985 Hubei genotype C strain (17) as well as the more recent Taiwanese strains (18,19).
One can only sum up from the above molecular virology data that there is no correlation between phylogenic evolution of EV71 in Asia and its supposed lethality and increased pathogenicity . The data do not support, or even argue against, the theoretical construct that there is an on-going genetic drift or evolution of EV71 towards increased virulence.
E Which is the Killer Virus?
Having read the basic facts, can you pinpoint the killer virus? Charles Calisher, an internationally known virologist, seems to think that the "conundrum between EV71 and adenovirus" cannot be settled yet (19). The following is an attempt on my part to answer the question. Just as in Rashomon, three different scenarios (hypotheses) could be conceived to re-construct the way in which the culprit virus killed.
Hypothesis I . This hypothesis takes off from the data, as they are perceived by the respective scientists and doctors in the three territories, at face value.
The gist of the Hypothesis is that both a strain of adenovirus and EV71 were killers in the two epidemics. Adenovirus ravaged the Sarawakian children, while EV71 killed them in Peninsular Malaysia and Taiwan.
Hypothesis II Both Hypotheses II and III presume a common culprit virus for both epidemics. They differ only in the identity of the culprit virus.
Hypothesis II thinks that enterovirus 71, an RNA virus of the Picornaviridae family, seems to be evolving towards a more virulent character. It struck the populace of both Sarawak and Peninsular Malaysia in 1997, together with other enterovirus and a few incidental viruses (eg. adenovirus, ECHO, Japanese encephalitis ), resulting in an unusually large outbreaks of hand, foot and mouth disease among children under the age of 5. On the crest of the outbreaks, it killed over 34 young children in Sarawak and at least 4 in Kuala Lumpur mainly by serious encephalomyelitis and its consequence - rapidly developing neurogenic pulmonary edema. In the following year's summer months, EV71 led a motley of other enteroviruses to strike the children of Taiwan with a HFMD epidemic of gargantuan proportion. At the crest of the epidemic, hundreds of pediatric patients were admitted to hospitals with serious CNS complications of EV71 infections. The most serious of these died of rhomboncephalitis (damage to the vital center demonstrated on CT scan) and associated acute NPE.
In hypothesis II, there can be no doubt that EV71, an emerging RNA virus, was the culprit virus at hand.
Hypothesis III
In the middle of 1997, an overly large outbreak of hand, foot and mouth disease struck Sibu and other areas of Sarawak. Synergistic conditions must have been created, either in the environment or susceptible children, to enable a new strain of adenovirus 21, abetted by another virus (ECHO, EV71, etc) to strike down 34 young children. While virus-inflicted acute myocardial failure was the probable cause of death, acute pulmonary edema and CNS damage could not be ruled out as mechanisms of death. A related secondary outbreak of HFMD in Peninsular Malaysia, the same virus probably also caused fatalities but was not detected because of its fastidious nature in culture.
While one of its phylogenetic cousin struck down a Singaporean child, the DNA virus named temporarily as SIBU97 or related adenoviruses, went on a rampage during the massive HFMD epidemic in Taiwan the following year. While over a million children in the island were afflicted with enteroviruses dominated by EV71 and Coxsackie A16, about 405 patients were hospitalized because of greater severity of their illnesses. 78 of these inpatients, where host factors were conducive, were killed by the emergent subgenus B adenovirus. The adenovirus was not identified because most virologist worked under the "EV71 paradigm" were not sufficiently thorough to culture it.
In this hypothesis, emergent strains of subgenus B adenoviruses, all closely related to SIBU97, were the killers. The adenovirus usually killed in partnership with another virus, usually an enterovirus but not always. The way in which the two viruses interacted to cause death by overwhelming the cardiopulmonary organs of the afflicted kids remains to be discovered.
F. Discussion
Hypothesis I , while being the safest way out for most scientists, is rather unsatisfactory because unifying features of two very similar epidemics are ignored.
EV71 or Adenovirus? Virological and molecular arguments
While it is difficult to apply strictly the nine factors that was mooted by Bradford Hill to assess causality in epidemics, some of them will be used to examine the issue on hand. These are: the consistency, specificity, strength of association, and congruence
(coherence) with scientific knowledge. While the isolation of the enterovirus 71 in large number of serious and fatal patients in the Taiwan epidemic supports Hypothesis II, causality needs to be scrutinized.
In terms of consistency, EV71 has shown a consistent pattern of causing a host of mild pathologies since 1969 in epidemics spanning Europe, US, Brazil, Australia. They had all manifested as hand-foort-and-mouth disease, herpangina and the more serious neurological signs of aseptic meningitis, encephalomyelitis and flaccid paralysis (20). The finding of "rhombencephalitis" was confined to the 1998 Taiwan epidemic while "neurogenic pulmonary edema" (cardiopulmonary failure) was described as a pattern only in the Taiwan and the 1997 Malaysian epidemics. Both pathologies were termed "unusual" by Dolin in NEJM (20). The point is, as with all emergent and reemergent disease, past consistency cannot be used as proof because these are supposedly signs of the virus making a genetic shift in virulence. It would erroneous in logic to allow previous epidemics with less virulent features to be used as evidence of consistency.
As for specificity, a whole motley crew of enteroviruses and one flavivirus were in the company of EV71 for both the Sarawakian and Taiwanese outbreaks. Specifity were especially lacking when Huang and associates admitted that serological testing for Japanese encephalitis (JE) were not done (21), in a country where pig-rearing is a common culture and JE used to be endemic! Strength of association is not particularly strong when you consider that most of EV71 isolated were from patients' throats or stool, not from the CSF (3). Only a few cases in Taiwan (4,6), 2 cases from Sarawak (1) and 4 cases in University Hospital, University of Malaya had positive EV71 identification from CNS sources (12).
When we consider the coherence of scientific evidence, the molecular virology work from CDC’s enterovirus laboratory practically argued against the hypothesis of emergence of new pathogenic strains of EV71. The tracing of genomic lineages of strains of EV71 from Sawarak, Taiwan and Peninsular Malaysia had placed the respective strains in either different clusters or even separate genotypes (in virological terms, the equivalent of the human "races") altogether. Betty Brown and Mark Pallansch, the virologists who have worked and published extensively on EV71, concluded that their phylogenetic studies on the virus had found no links between the three outbreaks (18). To accept Hypothesis II, one would need to assume that three non-related clusters of EV71 genetically shifted to a common pattern of virulence within a span of one to two year , inflicting very similar neuropathogenicity in the three places.
Coming to Hypothesis III, the strength of association is embodied mainly in the virological evidence of Cardosa and associates’ Lancet paper. Adenovirus was cultured from 11 of the clinical samples in the fatal cases from Sibu (compared with 4 enteroviruses). Three of these were from the heart, three from the CSF, one each from the brain and lungs (1).
However, to accept the Hypothesis we would need to explain why virologists in Peninsular Malaysia and Taiwan had not come across the adenovirus.
First of all, most virologists in this part of the world work under the mindset that all HFMD-associated deaths and neurological signs have EV71 lurking as the main culprit. I have labeled this as the "EV71-paradigm" effect. The paradigm probably originated unwittingly from CDC, with their quite legitimate concern about under-diagnosis of the rogue virus (11) . From personal observations, very few medical scientists working in the region have managed to free himself/herself from the paradigm. So most of the scientists end up asking the loaded question, "Is this an EV71 outbreak?". Very few ask the EV71-paradigm-free question, "What is the culprit virus responsible for fatalities in this outbreak?" It was with such a paradigm-free mindset that Jane Cardosa was able to discover the new strain of adenovirus.
The main reason for missing out on the Sibu97 was that it is a very fastidious virus to grow. The right culture systems and conditions as well as persistence with multiple passages before cytopathic effects can be seen. It grows preferably in human adenocarcinoma cells (A547 cells). A glance of the Taiwan papers’ methodology will confirm that not all the virology laboratories used this particular cell line. A recent letter sent to the Lancet by AbuBakar, Shafee and Chee (22) bore witness to the fastidious nature of the virus. The authors revealed that a similar adenovirus had been grown in Vero cells in Universiti Malaya but difficulty was encountered in propagating the virus. They had actually grown the adenovirus in 6 other Peninsular patients with HFMD, one having died of EV71-associated brainstem encephalomylelits.
NPE as a mechanism of death
'Neurogenic pulmonary edema' (NPE) had been a frequently invoked pathophysiological mechanism of death (3,4,6,10). One of the more insightful reviews, written by Stephen R. Ell, was of the opinion that the criteria for clinical and radiological diagnosis of the condition were rather unsatisfactory (23). There were no uniform criteria nor boundaries for both the diagnosis and exclusion of the condition. A rapid scan of the literature reveals that there are numerous causes of pulmonary edema: left ventricular failure, head injury, subarachnoid hemorrahge, raised intracranial pressure, bicuculline-induced status epilepticus, intrathecally-injected endothelins, poisonings by inhalation, hydrochloric acid instilled to the lungs, high concentration nitric oxide and others.
The experimental model most confluent with Hypothesis II was that developed by HI Chen (24) by inducing "intracranial hypertension" in dogs. The resultant central sympathetic activation caused extreme vasoconstriction in both the systemic and pulmonary resistance vessels. This then brought about acute left ventricular failure and pulmonary edema. In the Taiwan epidemic, it had been observed that there were signs of intense sympathetic nervous activity during episodes of EV71-associated pulmonary edema: tachycardia, hyperglycemia, paralytic ileus, neurogenic bladder, panic and startle reflex (3). The difficulty of establishing the mechanism lies also in proving cause and effect. Sympathetic discharge could have been brought on by primary cardiogenic shock. The adrenal medulla would then be a source of circulating catecholamines. More aggressive investigative measures should be adopted in future cases to establish or disprove NPE as a mechanism. This is because if excess sympathetic discharge can be proven, clonidine could then be used to reduce central sympathetic discharges. This would be potentially life-saving.
Since the pathogenesis of NPE could be triggered at so many levels, the connection with the hindbrain pathology would not be inevitable, even when the hindbrain was evidently damaged. The lungs, with their tachykinins-impregnated afferent nerves, are virtual minefields of inflammatory mediators most ready to affect cellular permeability (25, 26). Such direct and more accessible triggers of pulmonary edema should be ruled out before more distant and indirect causes are to be invoked. An interesting illustration of this point cropped up in Argentina, where a subgenus B adenovirus was found to cause death in an infant via Reye's syndrome and bronchiolitis obliterans (34). I think it may be fruitful in the future for virologists and experimental pathologists to explore the possibility of viruses taking advantage of the veritable arsenal of inflammatory mediators in the lungs.
Does the epidemiology support the "EV71 paradigm"?
As for the Taiwan epidemic, there are baffling features which should caution against the assumption that EV71 was the main culprit responsible for both the HFMD and deaths. The baffling disporportionality of small number of deaths when compared with the over million cases of lightly affected HFMD cases was mentioned in Section B. Furthermore, Monto Ho and colleagues (5) raised the question as to why such a large scale epidemic should come to visit the young of a population who already had half of its adults possessing antibodies against EV71. While it is to be expected that non-immune children would be susceptible to the pathogen when the adult population were immune, the authors suggested the possibility of importation of a more virulent strain. This possibility, at least from the direction of the two Malaysian outbreaks, has been rendered untenable by the phylogenetic work done by Pallansch, Brown and colleagues (16, 18).
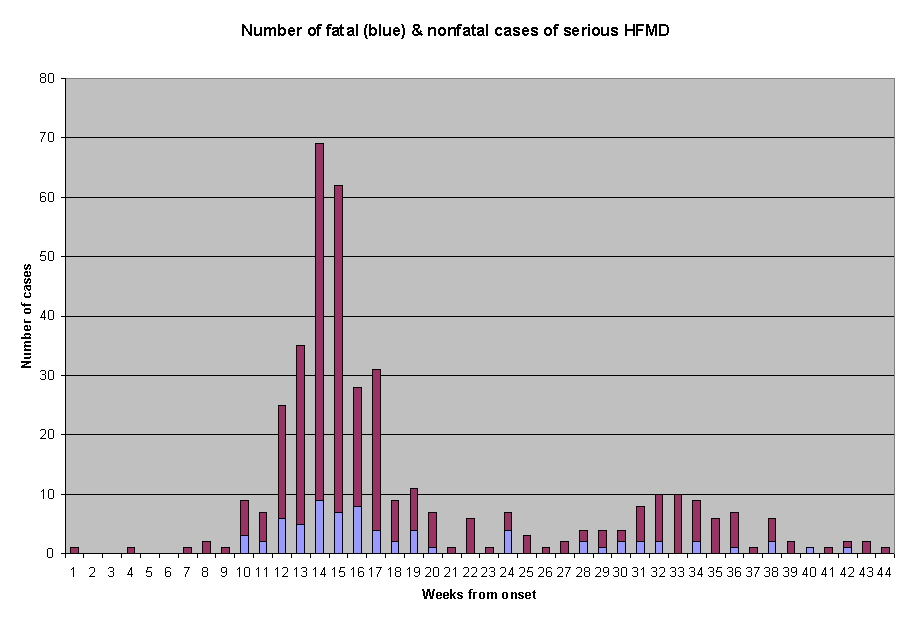
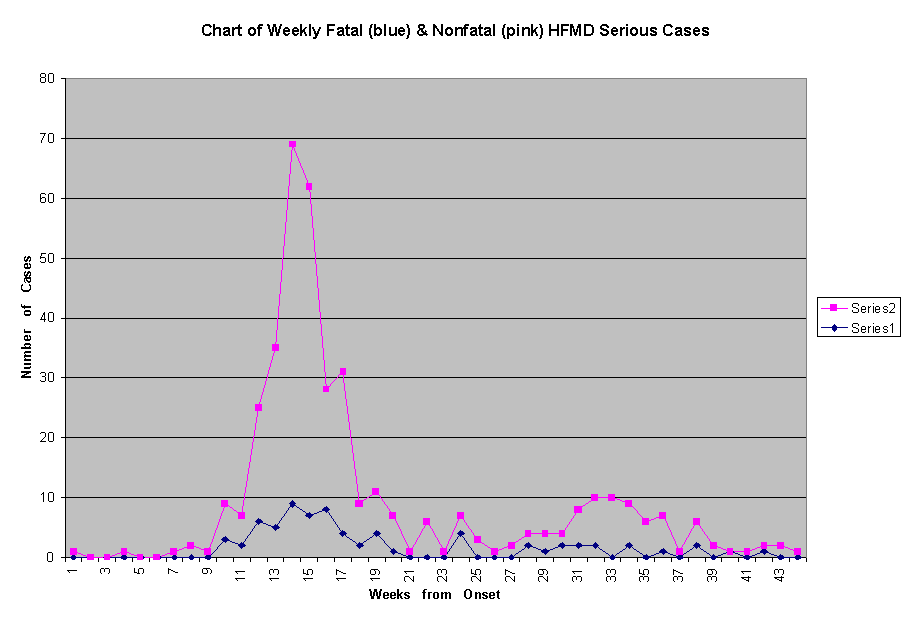
Ho and collegues were also unable to explain the difference in fatality rates among the severe HFMD cases in various regions of Taiwan. The Regions named as Central, Northern, Southern and Eastern showed fatality rates as varied as 31, 17.4, 13.6 and 7.7 percents respectively (5). Internet newpapers (Chinese language) during the epidemic had quoted Taiwanese doctors as saying that while cases in the Northern Region showed more neurological pathologies, the Central Region manifested more of the "myocarditis" type of deaths. I have further extracted data from their Figure 2 which showed the weekly chart of fatal and nonfatal cases of serious 405 HFMD admitted to 10 major hospitals. A re-plot of the figures as two concurrent linear series is shown in Figure 1. On a glance, the morbidity and mortality chart of a single pathogen should be expected to show more proportionate number of deaths during both the peak period and the nadir of infection. Chi-square (goodness of fit) analysis of the two series from Week 4/19 to Week 7/5 yielded the probability of the two series coming from one population was less than 0.01.
Furthermore, Chang and colleagues' single hospital study of 154 EV71 cases in Central Taiwan (3) showed that the group succumbing to pulmonary edema plus CNS lesions (11 cases all died) had much higher mortality rates when compared with the group with only CNS pathologies (only 2 out of 38 died or under-intensive care). Interestingly enough, the two groups demonstrated significant difference in sex ratios (male/female): 0.83 in the pulmonary edema group compared to 1.71 in the CNS involvement group. The latter conforms more with the generally observed sex ratios of enterovirus infections: 1.5 to 2.5: 1 (male : female) (27).
While all these could not amount to any proof of a second more lethal virus at work, Hypothesis III would be one scenario that was consistent with these "baffling" features of the Taiwan epidemic. According to Hypothesis III, the environmental and human conditions were such that in the mid-summer of 1998 to enable a huge epidemic of HFMD. This had resulted in the admission of over 400 serious cases of the infection, most probably caused by a squad of EV71-led enteroviruses. Different set of conditions were required to enable a SIBU97-related adenovirus to inflict mortal blows, probably "virus-induced myocardial akinesia", on a small number of these enterovirus-affected children. The conditions required for the main epidemic differ considerably from those of the fatal cases, giving rise to the above epidemiological anomalies.
The controversy over "myocarditis"
Lastly I shall discuss the issues centering around adenovirus-associated myocardial dysfunction. In a letter addressed to Lancet, Lucy Lum and colleagues had cited the absence of myocarditis pattern in histopathology of the 4 EV71-associated encephalomyelitis deaths as evidence against adenovirus involvement (28). They would have been arguing from the requirement of the Dallas Criteria, enacted by 8 American pathologists in 1984, of finding histopathological features of inflammatory infiltrate of the myocardium with non-infarct necrosis, in order to arrive at the diagnosis of "myocarditis" (29). One would have thought with advent of PCR and more molecular techniques in recent years, the Dallas criteria should not be regarded as all encompassing. Martin and the Towbin group demonstrated a peculiarity of adenovirus-associated myocardial pathology was that no sign of inflammation nor cellular infiltration was seen (30). Even when the new adenovirus was cultured from children's' heart samples in the Sarawak outbreak, absolutely no cellular infiltration was seen in the samples (1). Perhaps, the Dallas criteria and its diagnosis of "myocarditis" should be maintained as such, but any acute functional myocardial failure inflicted by a virus without histological signs of inflammation could be more appropriately described as "virus-induced myocardial akinesia".
A second argument cited against adenovirus-associated primary myocarditis as the main pathogenesis was the refractoriness of the moribund patients of Lum et al (28) to inotropic drugs. The counter argument would be that heart function could be adversely affected at more than one level. Most inotropic agents act either through the modulatory function of the adenyl cyclase-cyclic AMP mechanism or the global storage of calcium within the sarcoplasmic reticulum (31). While we are ignorant of the mechanism of virus-induced myocardial dysfunction, other levels of myocardial function could well be the vulnerable points through which viruses act. For example the ryanodin receptor channels pivotal for electrical-contraction coupling (32) could be such a vulnerable point.
An interesting question arose from one of the epidemiological features of the Taiwan epidemic. Monto Ho and colleagues (5) mentioned that, among the hospitalized HFMD cases, the 7 to 12-month-old had a significantly higher fatality rate (43%) when compared with that of the 6-months old age group (16%). An idea that immediately sprang to mind, was that,"Could the contrasting difference in fatality be accounted for by developmental phenomena such as the delayed expression of virus receptors in infants?" Adenovirus were known to employ major histocompatibility complex (MHC) molecules as receptors (35). Thus more information on the developmental expression of either MHC molecules or the common CVB & adenovirus receptor might throw light on the differential fatality rates during infancy (working within the Hypothesis III paradigm).
While evidence for virus interactions or synergism is rather scanty, report of finding both mumps virus with either CMV or enterovirus in the heart muscles of children with endocardial fibroelastosis was an early prelude of such interactions. (7). The more exciting of these was the finding of a common receptor in heart muscle for two carodiovirulent viruses - coxsackie B (CBV) and adenovirus subgroup C (10). This provides a shared mechanism by which two very contrasting viruses can invade the heart. The genome of an coxsackie virus is a single-strand RNA with 7.5 kb only; in contrast, that of adenovirus is a 35 kb double stranded DNA (10).
Viruses in general have strategies to evade immune responses directed against them. For example, homologues of chemokines might be encoded by viruses in order to block cytotoxic T lymphocytes from recruitment and amplification of further effector cells, thus thwarting the virus-provoked immune and inflammatory response (33). It is therefore logical to postulate that immune evasion mechanism of one virus might enable another virus to gain a foothold in cells (and to become cardiovirulent). Price and colleagues further argued that since viruses co-evolve with human hosts, it would not be surprising to find two or more viruses converging towards common human targets in a synergistic way (33).
Summary
I have summarized key points of clinical, epidemiological, virological and molecular data from published literature on the two HFMD-associated fatal epidemics that had ravaged both young children in both Malaysia and Taiwan. By way of forming hypotheses, I have attempted to reenact the common features of the 1997 Malaysian and 1998 Taiwan epidemics into events involving a single virus with emerging pathogenicity.
Hypothesis II postulating EV71 as the main culprit virus is argued to be untenable because of differing phylogenetic origins of the isolated EV71 from patients of the three territories (Sarawak, Peninsular Malaysia and Taiwan). Furthermore, the gross proportion of the Taiwanese lightly infected HFMD : serious HFMD : fatal cases were estimated to be 1.48 million : 327 : 78; while the comparable approximate proportion of the Sarawak cases were 10,000 : ? : 34. Such disproportionately small number of fatalities (when compared with the Sarawakian estimate) as well as other examined anomalous features of the Taiwan epidemic could be better explained by presuming an undiscovered fatal pathogen in the midst of gargantuan epidemic of light HFMD caused by EV71 and other enteroviruses.
The bulk of the the virological and molecular evidence supporting the Hypothesis III was published in a singular paper in Lancet by Cardosa and colleagues in September 1999. The evidence pointed towards a new subgenus B adenovirus named temporarily Sibu97 as the main culprit virus in the Sarawak outbreak, perhaps interacting with enteroviruses. The pervasive influence of the "EV71 paradigm" and the fastidious nature of the new adenovirus could have accounted for failure of virus laboratories outside of Sarawak’s UNIMAS to culture and characterize it. All the cumulative evidence were consistent with the hypothesis that Sibu97 or its close relatives was the undiscovered lethal pathogen in both the Taiwan and Peninisular Malaysian outbreaks.
In the Discussion, I further argues that adenovirus-associated "myocardial akinesia" is scientifically a more plausible mechanism of death than the EV71-associated rhombencephalitis and "neurogenic pulmonary edema". The possibility of two viruses interacting with each other through their co-evolution with human genomes, especially in immune evasion, in order to achieve common pathogenicity is raised.
Acknowledgment
Dr.Phua Kai Lit for running the Chi-square test.
References
- Cardosa MJ, Krishnan S, Tio PH, Perera D, Wong SC. Isolation of subgenus B adenovirus during a fatal outbreak of enterovirus 71-associated hand, foot, and mouth disease in Sibu, Sarawak. Lancet 1999; 354: 987-991.
- Tan Poh Tin. Clinical presentation of 27 deaths in Sarawak 14 April - 22 June 1997. Under 'Enterovirus 71 epidemic, children: Clinical Signs'. ProMED-mail post 19 June 1998.
- Chang LY, Lin TY, Hsu KH, Huang YC, Lin KL, Hsueh C, Shih SR, Ning HC, Hwang MS, Lee CY. Clinical features and risk factors of pulmonary oedema after Enterovirus-71-related hand, foot, and mouth disease. Lancet 1999; 354 (9191): 1682 - 1686.
- Huang CC, Liu CC, Chang YC, Chen CY, Wang ST, Yeh TF. Neurologic complications in children with enterovirus 71 infection. NEJM 1999; 341: (13) 936 - 942.
- Ho M, Chen ER, Hsu KH, Twu SJ, Chen KT, Tsai SF, Wang JR, Shih SR. An epidemic of enterovirus 71 infection in Taiwan. NEJM 1999; 341: (13) 929 - 935.
- Chang LY, Huang YC, Lin TY. Fulminant neurogenic pulmonary edema with hand, foot and mouth disease. Lancet 1998; 352: 367-368.
- Bowles NE, Towbin JA. Viral heart muscle disease in children. www.unmc.edu/Pathology/myocarditis/bowtow.html 1996.
- Huber SA, Gauntt CJ, Sakkinen P. Enteroviruses and mycarditis: viral pathogenesis through replication, cytokine induction, and immunopathogenicity. In Advances in Virus Research, ed. Maramorasch K, Murphy FA, and Shatkin AJ, Academic Press, San Diego 1999; 51: 35-80.
- Andreoletti L, Hober D, Decoene C et al. Detection of enteroviral RNA by polymerase chain reaction in endomyocardial tissue of patents with chronic cardiac diseases. J Med Virol 1996; 48: 53-9.
- Bowles NE & Towbin JA. Molecular aspects of myocarditis. Curr Opinion Cardiology 1998 13: 179-184.
- Alexander JP Jr, Baden L, Pallansch MA, Anderson LJ. Enterovirus 71 infections and neurologic disease - United States, 1977 - 1991. J Infect Dis 1994; 169: 905 - 908.
- Lum CS Lucy, Wong KT, Lam SK, Chua KB, Goh AYT, Lim WL, Ong BB, Paul G, AbuBakar S, Lambert M. Fatal enterovirus 71 encephlomyelitis. J. Pediatr 1998; 133: 178 - 795.
- AbuBakar S, Chee HY, Shafee N, Chua KB, Lam SK. Molecular detection of Enteroviruses from an outbreak of hand, foot and mouth disease in Malaysia in Scand J Infect Dis 1999; 31 (4): 331-5.
- Baker AB. Poliomyelitis: a study of pulmonary edema. Neurology 1957; 7: 743 - 751.
- Cardosa MJ, Institute of Health Community Medicine, UNIMAS, Kuching. Personal communication.
- AbuBakar S, Chee HY, Al-Kobaisi MF, Xiaoshan J, Chua KB, Lam SK. Identification of enterovirus 71 isolates from an outbreak of hand, foot and mouth disease with fatal cases of encephalomyelitis in Malaysia. Virus Res 1999; 61(1): 1-9.
- Brown BA, Oberste MS, Alexander Jr JP, Kennett ML, Pallansch M: Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. J Virology 1999; 73(12): 9969 - 75.
- Pallansch MA, Brown BA. Division of Viral & Rickettsial Diseases, Centers for Disease Control and Prevention, Atlanta, Georgia, USA. Personal communication over e-mail.
- Calisher CH. Enterovirus 71 Asia ProMED-mail post 26.12.1999
- Dolin R. Enterovirus 71 - Emerging infections and emerging questions. NEJM 1999; 341: 984-985.
- Huang CC, Liu CC, Chang YC. Letter to Editor. NEJM 2000; 342: (5) 357.
- AbuBakar S, Shafee N, Chee HY. Adenovirus in EV71-associated hand, foot, and mouth disease. Correspondence in Lancet 2000; 355(9198):
- Ell, SR. Neurogenic pulmonary edema: a review of the of the literature and perspective. Investigative Radiology 1990; 26: 499-506.
- Chen HI. Hemodynamic mechanisms of neurogenic pulmonary edema. Biol Signals 1995; 4: 186-92.
- Barnes, PJ, Chung KF, Page CP. Inflammatory mediators of asthma: an update.Pharmacol Review 1998; 50(4): 515-60.
- Germonpre PR, Joos GF, Pauwels RA. Characterization of the neurogenic plasma extravasation in the airways. Arch Int Pharmacodyn Ther 1995; 329: 185 – 203.
- Jubelt B. Other enteroviral diseases. In ‘Handbook of Neurovirology’, ed. McKendall RR & Stroop WG. 1994:503-519.
- Lum LCS, Wong KT, Lam SK, Chua KB, Goh AYT. Adenovirus in EV71- associated hand, foot, and mouth disease. Correspondence in Lancet 2000; 355: 146.
- Aretz HT, Billingham ME, Edwards WD, Factor SM, Fallon JT, Fenoglio Jr. JJ, Olsen EGJ, Schoen FJ. Myocarditis: a histopathologic definition and classification. Am J Cardiovas Pathol 1987; 1: 3-14.
- Martin AB, Webber S, Fricker FJ, Jaffe R, Demmler G, Kearney D, Zhang Y-H, Bodurtha J, Gelb B, Ni J, Bricker T, Towbin JA. Acute myocarditis: rapid diagnosis by PCR in children. Circulation 1994; 90: 330-339.
- Bristow, MG. Why does the myocardium fail? Insights from basic science. Lancet 1999; 352 (suppl 1): 8-14.
- Cannel MB, Soeller C. Sparks of interest in cardiac excitation-contraction coupling. Trends in Pharmacol Sc 1998; 19: 16 - 20.
- Price, DA, Klenerman P, Booth BL, Phillips RE, Sewell AK. Cytotoxic T lymphocytes, chemokines and antiviral immunityu. Immunology Today 1999; 20(5): 212-6.
- Mistcenko AS, Robaldo JF, Guiterrez R. Fatal adenovirus infection associated with new genome type. J Med Virol 1998; 54: 233-6.
- Lentz TL. Viruses as ligands of eukaryotic cell surface molecules. In 'Molecular basis of virus evolution'. Ed. Gibbs AJ, Calisher CH, Garcia- Arenal F. 1995 Cambridge University Press.
Back to Vads Corner
Back to Cybermed Articles